Predicting the conformational flexibility of antibody and T cell receptor complementarity-determining regions
Many proteins are flexible molecules that adopt several stable structures, termed conformations, and transitions between them can be fundamental for their function1,2. Antibodies and T cell receptors (TCRs) primarily engage their targets through six loop motifs called the complementarity-determining regions (CDRs)3. Structural flexibility of CDRs has been linked to several key functional properties. For some antibodies, conformational changes are known to be required for antigen recognition4. The ability of an antigen receptor to adopt multiple conformations has also been associated with polyspecificity as different structural states allow for greater variability in the recognized antigens5,6. Furthermore, flexibility has an effect on binding affinity as it directly impacts the entropic costs of antigen binding7 and rigidification has been observed as one of the natural mechanisms used to increase affinity8,9.
Specificity and affinity are two critical properties of antibody and TCR therapeutics. To maximize target interactions and minimize off-target interactions, a therapeutic should have high affinity and specificity10. This suggests that therapeutics should have a preference towards rigidity. However, there is some evidence indicating that conformational flexibility allows a better recognition of mutated antigen variants and could be desired when designing broadly neutralizing antibodies11,12. In either case, a method predicting CDR flexibility would allow both an enhanced investigation of antibody function and the potential to tune the desired therapeutic properties.
Computational predictions of a single, static structure of a protein from its sequence is now considered a routine task (for example, refs. 13,14,15), and more recently developed tools are also showing promise at protein complex prediction (for example, refs. 16,17). However, predicting structures of more than one conformational state remains challenging. One factor that has limited progress in conformation prediction is the scarcity of suitable data.
Evidence on conformational flexibility can currently be obtained from several experimental sources. Nuclear magnetic resonance spectroscopy and hydrogen–deuterium exchange mass spectrometry can be used to measure the protein dynamics in solution, although they do not typically provide atomically resolved flexibility information (see ref. 18 for a review of nuclear magnetic resonance and ref. 19 for hydrogen–deuterium exchange methods). X-ray crystallography is the standard method used to obtain the high-resolution structures of conformational states. These can be captured by solving separate structures of the same protein under different conditions. Because multiple structures must be available, the number of proteins for which flexibility can be assessed from the crystallographic data is much smaller than the total number of solved structures. Crystal structures have been used to explore the flexibility of specific loop types20 or for case studies of full-length proteins21,22,23, but there has been little systematic mining of the Protein Data Bank (PDB) for all instances of the same sequence representing alternative conformations24.
Molecular dynamics (MD) simulations provide a computational way to generate conformational ensembles. MD simulations are computationally expensive, and therefore, even the largest databases of standardized MD simulations are not yet sufficient for training machine learning models25.
Despite these data challenges, a number of methods have been developed that attempt to predict structures of protein conformational ensembles. Recent approaches have concentrated on modifying the AlphaFold2 (AF2) inference procedure to increase the diversity of outputs (for example, refs. 22,23,26,27,28,29,30). These methods target multiple sequence alignment (MSA), one of the main inputs of AF2, from which co-evolutionary signals are extracted to infer protein residues probably in close proximity. In theory, the MSA should contain co-evolutionary information for all conformational states. Deconvolving these signals is generally attempted by reducing the depth of the MSA, for example, through random subsampling22 or sequence clustering26. More recently, a range of methods were specifically trained for the task of conformation prediction (for example, refs. 31,32,33,34,35,36,37). These generally take the form of generative protein structure prediction models trained on the PDB and a small number of MD simulations.
Detailed evaluation of conformation prediction tools is complicated by data scarcity, but available evidence suggests that reliable predictions are not yet possible. Many methods have only been evaluated on one or a few case studies, which may not accurately reflect their predictions across diverse sets of proteins21,28,29,30,33. The evaluation of some methods on slightly larger test sets show that the diversity of predicted structures is generally increased; however, the conformational landscape is not covered with high accuracy36. The prediction of the conformational states of antibody and TCR CDRs specifically has only been assessed on a handful of case studies21.
In this work, we focus on the flexibility of the functionally important antibody and TCR CDR3s. In an attempt to address the issue of data scarcity, we consider loop motifs with the same secondary structure pattern, defined as loops bounded by two antiparallel β-strands, across all proteins. Through a systematic mining of the PDB38 and antibody- and TCR-specific databases39,40, we created antibody-like loop conformations (ALL-conformations)—a dataset containing 1.2 million crystal structures of loops with 100,000 unique sequences. The dataset captures all the experimentally observed conformations of loop motifs found between pairs of antiparallel β-strands including both antibody and TCR CDR3s (Fig. 1a). We analysed the structural flexibility in the ALL-conformations set and label more than 20,000 unique loop sequences by their ability to undergo conformational changes (Fig. 1b). Using these data, we built an Immunoglobulins and TCRsFlexibility classifier (ITsFlexible)—a method that classifies whether CDR3s are rigid (adopt a single conformation) or exhibit flexibility (transition between multiple states) (Fig. 1c). ITsFlexible predicts the flexibility of CDR3s evaluated in ensembles of crystal structures with high accuracy and achieves state-of-the-art performance. The model also effectively generalizes to a test set derived from MD simulations. Furthermore, we used ITsFlexible to predict the flexibility of three CDRH3 loops with no solved structures and used cryogenic electron microscopy (cryo-EM) to experimentally determine their conformations. These experiments showed that two of the three model predictions were correct.
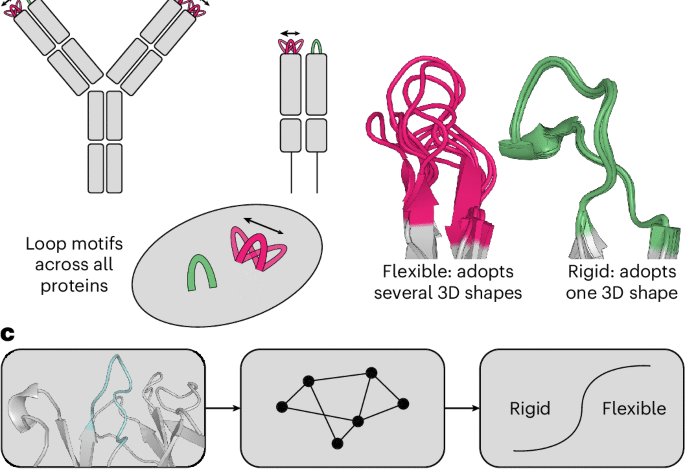
a, ALL-conformations is a dataset that contains the crystal structures of antibody CDR3s, TCR CDR3s and CDR-like loop motifs across all proteins. The dataset captures all the observed conformational states of such loops. b, Loops are labelled as either flexible (if they are observed in more than one conformation) or rigid (if evidence suggests that they adopt a single conformation). We define a conformation by structural similarity and use an RMSD of 1.25 Å as a threshold to separate states. c, Flowchart detailing the ITsFlexible method predicting the conformational flexibility of CDR loops. The structure and sequence of a loop (cyan) and its context (grey) are extracted from a PDB file, and a graph representation is generated. A graph neural network (GNN) classifies loops as conformationally flexible or rigid.
ALL-conformations is released on Zenodo (https://doi.org/10.5281/zenodo.15784241)41, and ITsFlexible is available on GitHub (https://github.com/oxpig/ITsFlexible) and Zenodo (https://doi.org/10.5281/zenodo.16891380)42.
Results
ALL-conformations dataset
ALL-conformations is a dataset that captures the conformational flexibility of loop motifs bounded by two antiparallel β-strands, which included five subsets, antibody CDRH3s and CDRL3s, TCR CDRB3s and CDRA3s and loop motifs across all proteins in the PDB (Supplementary Fig. 4). The dataset collects available crystal structures of such loops and contains the structures of all experimentally observed conformational states.
Extracting CDR3 structures from structural antibody database (SAbDab)39 and structural T cell receptor database40 and loop motifs from the PDB38, we obtain a total of more than 1.2 million examples with more than 100,000 unique sequences (Table 1). The PDB set contains loops between 1 and 87 residues; however, it is heavily enriched for shorter loops (Supplementary Fig. 5). Length distributions are different for CDR3s with a peak of around 10 to 15 residues and no loops shorter than 4 amino acids (Supplementary Fig. 6). We found that a large number of loop sequences are represented by multiple crystal structures (Supplementary Figs. 5 and 6).
The loops were classified as rigid, flexible or unknown (Methods). Loops for which multiple conformations are observed in the crystal structures were labelled as flexible. Here we define a conformation as a cluster in which the pairwise root mean square deviation (RMSD) of any member is below a threshold of 1.25 Å. This approach was chosen because it was previously shown to provide a good functional clustering of antibodies43. A conformation should, therefore, reflect a functionally distinct structural state. It is not possible to guarantee that all loops only observed in a single conformation are indeed rigid as there always remains a possibility that additional conformational states have not yet been captured. To address this issue, we only label loops as rigid if they adopt the same conformation in more than five structures. This should ensure that the set is heavily enriched for loops with a single accessible conformation. Using these definitions, we identified more than 16,000 rigid and 4,000 flexible loops (Supplementary Table 5). For the evaluation of the impact of RMSD threshold on the proportion of two classes, see Supplementary Fig. 10 and Supplementary Section 2.3.
Predicting the flexibility of CDR-like protein loop motifs
The ALL-conformations set was used to train ITsFlexible, a model that predicts protein loop flexibility. ITsFlexible is a graph neural network (Supplementary Fig. 1) that binary classifies loops if they can occupy multiple conformations (flexible) or adopt a single stable state (rigid) from inputs encoding the sequence and structure of a loop and its structural context (Methods).
We trained and evaluated ITsFlexible on the PDB set of ALL-conformations, containing loop motifs observed across any protein. The data split was performed based on sequence identity and test set loops had a maximum of 80% sequence identity with length-matched loops in the training and validation sets. Classifier performance was compared with random classification, three baseline models and a zero-shot flexibility prediction workflow based on AF2 predicted local distance difference test (pLDDT)13 (Fig. 2). Random classification shows validation metrics when randomly classifying a dataset with the given proportion of labels. The baselines predict flexibility from biophysical features such as loop length and solvent exposure, which have previously been shown to influence loop dynamics20,44. Long loops contain more bonds around which they can rotate and solvent exposure reduces steric hindrance, which restricts conformational rearrangements. pLDDT is a residue-level confidence score and provides a good indicator of disordered protein regions13. As the test set contains several structures containing each loop, an ITsFlexible prediction was made for all of them and the metrics were calculated based on the mean prediction score.

a,b, Classification on the PDB test set containing 2,845 loop motifs is evaluated with metrics of PR AUC (a) and ROC AUC (b). ITsFlexible performance (light grey) is compared with random classification (red), three biophysical baselines (green), an AF2 pLDDT-based model (blue) and two ITsFlexible versions with input ablations (dark grey). Exact PR AUC and ROC AUC values are presented in Supplementary Tables 9 and 10.
ITsFlexible is able to classify loop flexibility (area under the precision–recall curve (PR AUC) of 0.62 and receiver operating characteristic area under the curve (ROC AUC) of 0.84) and outperforms our baselines and zero-shot classifier. We investigated features predictive of loop flexibility through the ablation of model inputs. ITsFlexible-loop is similar to our default model but inputs are reduced to the structure and sequence of only the loop itself (Supplementary Section 1.1). ITsFlexible-sequence is a convolutional-neural-network-based model trained on a sequence encoding loops (Supplementary Section 1.1). ITsFlexible-loop and ITsFlexible-sequence achieve similar performance. Both outperform the relevant baseline (only baseline length is relevant as solvent exposure depends on the structural context, which both models do not consider) but are less predictive than ITsFlexible (Fig. 2).
These results give some indication of the factors that determine the conformational flexibility of protein loops. It is well known that longer and solvent-exposed loops tend to be more flexible than shorter and buried ones20,44. Although our findings agree with these general trends, we also show that the loop sequence is more predictive of flexibility than length alone, suggesting that the sequence of a loop impacts its ability to adopt multiple conformations. A large boost in performance was achieved by encoding the structural context of the loop motif. In line with previous evidence from MD studies44, this highlights that the interactions of a loop with its context within the protein are important determinants of its conformational dynamics.
ITsFlexible is highly predictive of CDR3 flexibility
We next investigated the ability of ITsFlexible, trained on general proteins loops, to predict the flexibility of antibody and TCR CDR3s. The model was evaluated on the CDR3 sets of ALL-conformations, which were designed to have no overlap, defined as more than 80% aligned sequence identity, with the training and validation sets.
We initially evaluated ITsFlexible using crystal structures as the input. In addition to the baselines and the pLDDT-based predictor introduced in the previous section, we evaluated two additional zero-shot flexibility predictors. The first workflow predicts flexibility based on the diversity in CDRs modelled by multiple AF2 runs with subsampled MSAs (Methods). The second workflows uses the residue-level confidence score (root mean square predicted error (RMSPE)) of ABodyBuilder2 (ABB2), an antibody-specific structure predictor45.
ITsFlexible proved to be highly predictive of antibody and TCR CDR3 flexibility (Fig. 3). Our method outperforms the biophysical baselines and zero-shot classifiers in nearly all CDR test sets. The only exception is the CDRA3 set in which the MSA subsampling approach surpasses ITsFlexible by a narrow margin. However, ITsFlexible substantially outperforms all other methods on CDRH3s, the largest and, therefore, the most representative set, and is the only method that consistently achieves high predictive accuracy across all four test sets. We also point out that ITsFlexible was evaluated with the most stringent train–test split compared with the zero-shot models. Specifically, CDRs in the ITsFlexible training set were filtered by 80% sequence identity to the test sets, whereas 100% sequence identity was used for ABB2 and the AF2 training set may even contain test set overlaps.

a,b, Classification on the four test sets is evaluated with metrics of PR AUC (a) and ROC AUC (b). ITsFlexible performance from inputs of crystal structures, IB and AF2 models (light grey) is compared with random classification (red), three biophysical baselines (green) and three zero-shot models based on the outputs of protein structure prediction tools (blue). Exact PR AUC and ROC AUC values are presented in Supplementary Tables 9 and 10. c,d, Example of antibodies predicted to be flexible and rigid by ITsFlexible. c, Overlay of six structures of the same antibody Fv with CDRH3 predicted to be flexible. Crystal structures indicated that the CDRH3 (highlighted in colour) adopts three different conformations (red, blue and green). d, Overlay of 22 structures of the same antibody Fv with CDRH3 predicted to be rigid. The CDRH3 (red) occupies the same conformations in all 22 structures.
We conducted a more detailed investigation of the ABB2 confidence score. This analysis revealed that RMSPE is potentially more indicative of the number of times a certain antibody occurs in the training data rather than flexibility (Supplementary Section 2.4).
Comparing the classification performance between the four CDR sets (using ROC AUC), we observed the best results for L3s and B3s with slightly lower values for H3s and A3s. Antibody H3s have higher sequence diversity than L3s, which is expected to make classification more difficult. The lower performance for A3s could be linked to the findings of a different pattern of genetic diversity compared with B3s46. However, we also highlight the small size of the A3 set, which may result in a less accurate estimation of performance.
Furthermore, we analysed the consistency of ITsFlexible predictions when using different structures containing the same loop as the model input. The results show that variations in prediction scores are generally small, demonstrating consistency (Supplementary Fig. 7).
As structural data are only available for a subset of known antigen receptor sequences47,48, ITsFlexible was additionally tested using predicted structural models from ImmueBuilder (IB) and AF2 as inputs. On the antibody CDR sets, similar performance is observed for predictions from crystal structures and IB models and slightly worse predictions are made from AF2 models. Although there is no clear correlation between model quality and the error in the ITsFlexible prediction, we observed that higher prediction errors tend to occur at lower model quality (Supplementary Fig. 8). The difference in classification performance may, therefore, be tied to the lower quality of AF2 compared with IB models of our test set (Supplementary Table 7). On the TCR sets, predictions from AF2 models are marginally better in PR AUC than from IB models with a larger difference in ROC AUC. As for the antibody sets, we observed a lower model accuracy and a larger error in ITsFlexible prediction score for AF2 (Supplementary Table 7). However, for TCRs, this does not deteriorate the classification metrics.
Throughout the analysis in this paper, we made the following assumptions to determine CDR flexibility. CDR3s are defined as International ImMunoGeneTics (IMGT) residues 107–116 (Methods and Supplementary Fig. 4), structural diversity is calculated as the loop RMSD after alignment on the loop residues and a conformation is defined as a structural cluster with pairwise RMSD between any member below 1.25 Å. Additional analysis was performed on datasets with CDR3s defined by their exact secondary structure (Supplementary Table 8), flexibility calculated by alignment on the framework residues (Supplementary Fig. 9) and different choices of RMSD thresholds (Supplementary Fig. 10). We found that ITsFlexible’s high predictive accuracy remains qualitatively similar for the first two experiments, whereas the choice of threshold impacts classification to a minor extent.
ITsFlexible matches CDR flexibility in MD simulations
Although crystallographic data can reveal the conformational states of CDRs, it does not directly measure flexibility, and there always remains the possibility that additional conformational states exist but have not been captured. A limitation of the ALL-conformation dataset, therefore, is the lower confidence of rigid compared with flexible labels. Although we introduced the requirements to address this limitation (Methods), the true extent of flexibility may well be underestimated.
We used MD simulations to classify CDR flexibility by simulating a set of 19 antibodies and labelling the flexibility of CDR3s (Supplementary Table 2). As expected, we observed a higher proportion of flexible CDR3s in MD (84% for CDRH3 and 37% for CDRL3) than the crystal structure data (64% for CDRH3 and 18% for CDRL3). ITsFlexible achieved near-perfect separation for CDRH3s, with slightly worse performance for CDRL3s (Fig. 4), suggesting that ITsFlexible also detects signals promoting flexibility in physics-based molecular simulations.

a,b, Classification of CDRH3s and CDRL3s is evaluated with metrics of PR AUC (a) and ROC AUC (b). A prediction was made for each representative structure extracted from MD and classification performed based on the maximum ITsFlexible score observed across the ensemble. Exact PR AUC and ROC AUC values and performance based on the mean ITsFlexible score observed across the ensemble are presented in Supplementary Table 17.
Cryo-EM experiments confirm predicted flexibility
We chose three antibodies as challenging test cases for experimental validation using cryo-EM. Antibodies in the patent and literature antibody database49 against the influenza H1N1 hemagglutinin (HA) with ITsFlexible score below 0.1 (rigid examples) or above 0.5 (flexible examples) were considered. Three antibodies with low sequence identity to any examples in the training set and loop lengths opposing trends observed in ALL-conformations (long loop for rigid case and shorter loops for flexible cases) were selected. These were imaged in complex with the antigen and the heterogeneity in the density maps, indicative of conformational variety50, was analysed.
Antibody 1 is as an example of a long CDRH3 (length 19, longer than 86% of CDRH3s in ALL-conformation) predicted to be rigid with high confidence (ITsFlexible score of 0.02). The cryo-EM data revealed that the majority of good particles selected from two-dimensional classification adopt one homogeneous three-dimensional (3D) class, which was used to build a high-resolution consensus structure (Fig. 5). In agreement with model prediction, these data show that the CDR occupies a single conformation.

High-resolution structure of the three antibodies in complex with influenza H1N1 HA were solved. a,d,g, Top and side views of the cryo-EM map of antibodies (heavy chain, dark colour; light chain, bright colour) in complex with the antigen (grey). In each structure, three symmetrically arranged copies of the antibody were captured. Antibody 1 (9N5Y; a) binds to the HA stem and antibodies 2 (9N5Z; d) and 3 (g) to the HA head. b,e,h, Cartoon representation of antibody–antigen binding interfaces of antibodies 9N5Y (b), 9N5Z (e) and 3 (h). CDRH3s are shown in different shades of colour and binding interactions are highlighted in stick representation. c,f,i, Summary tables showing the CDRH3 length and sequence, sequence identity to the closest example in the ITsFlexible training set, the ITsFlexible prediction score (a higher value indicates a higher likelihood of being flexible), a binary prediction of loop flexibility based on the ITsFlexible score and the flexibility determined with the cryo-EM experiments for antibodies 9N5Y (c), 9N5Z (f) and 3 (i). Additional metadata for the three antibodies are shown in Supplementary Table 21.
Antibody 2 is an example with a slightly shorter CDRH3 loop (length 16) predicted to be flexible with high confidence (ITsFlexible score of 0.76). Our initial data processing showed high flexibility in the binding interface of the antibody and the antigen although at a low resolution. With further 3D classification and data collection, we were able to obtain a high-resolution structure of one of the binding states (Fig. 5). Data resolution did not allow us to build models of distinct conformational states, which makes it hard to localize the flexibility to the CDRH3 loop. However, the density maps show clear conformational heterogeneity at the binding interface and induced by the antibody (Supplementary Videos 1 and 2). Flexibility of CDRH3 is a likely cause for this observation.
Antibody 3 is a further example of a CDRH3 predicted to be flexible. The CDRH3 is even shorter than in antibody 2 (length 13) and the ITsFlexible score of 0.60 suggests a lower prediction confidence. The cryo-EM data disagreed with the prediction and revealed no heterogeneity (Fig. 5).
A potential limitation of the experimental setup is that antibodies were imaged in complex with the antigen. This choice was made as the minimum particle size required for cryo-EM complicates the study of free antibodies. Binding can rigidify residues as additional molecular interactions with the antigen impose extra constraints7, and we expect ITsFlexible predictions to better match the flexibility of the unbound antibody (see the ‘Discussion’ section). This limitation could be a reason for the absence of flexibility observed in experiments for antibody 3. In particular, we observe that a glutamate residue at the tip of the CDRH3 forms three hydrogen-bond interactions with the antigen (Fig. 5). Accordingly, an argument could also be made that the unbound state of antibody 1 is more flexible than observed. The data, nevertheless, provide clear evidence for reduced flexibility of antibody 1 compared with antibody 2 when imaged under the same conditions, matching our predictions.
Discussion
Conformational changes are fundamental to the functional properties of many proteins1,2. In antibodies and TCRs, flexibility—especially of the highly important CDR3s—impacts key properties such as affinity and specificity 5,6,7,8. However, predicting protein flexibility remains challenging, as current structure prediction tools struggle to capture multiple conformational states, particularly for antigen receptor CDRs21,36. One factor that has limited the progress in conformation prediction is the absence of large datasets necessary to train and evaluate such machine learning models.
Here we focused on the computational prediction of antigen receptor CDR3 flexibility. To help overcome the lack of data, we collected structural motifs with the same secondary structure pattern, defined as a loop connecting two consecutive antiparallel β-strands, across all proteins. Mining the PDB, we created ALL-conformations—a dataset containing more than 1.2 million crystal structures of such loops with more than 100,000 unique sequences. Analysing the conformational flexibility in ALL-conformations, we were able to label 20,000 loops with high confidence as either flexible (adopting multiple conformations) or rigid (occupying a single state).
Using the ALL-conformations dataset, we developed ITsFlexible—a method for the classification of CDR loops as either flexible or rigid. The model was trained on the subset of loop motifs from general proteins in the PDB and evaluated on its ability to predict the CDR flexibility in an out-of-distribution setting. ITsFlexible achieved state-of-the-art performance on crystal structure test sets and generalized effectively to the MD simulation data. In addition, we showed that the model achieves similar performance when using inputs of structural models rather than crystal structures, highlighting its applicability to antibodies without experimentally solved structures.
An ablation of ITsFlexible inputs indicated biophysical factors that influence the flexibility of CDRs. In line with previous MD studies44, we identified the arrangement of residues in the structural context of CDRs as a key factor driving flexibility.
Furthermore, our analysis showed that uncertainty scores of protein structure prediction tools and diversity in structural ensembles generated with established workflows are not reliable predictors of CDR flexibility. The AF2 pLDDT score has been described as a good indicator for disordered regions of proteins13; however, we found that it was not highly predictive of CDR flexibility. Similarly, the predicted error of the antibody-specific ABB2 correlates more strongly with the number of times a particular sequence was present in the training set than flexibility. We also assessed the diversity of CDR ensembles produced by AF2 MSA subsampling as a predictor of flexibility. MSA subsampling is a popular workflow to model multiple conformational states of proteins22,23,26,27,28,29,30. Although this approach is more predictive than confidence scores, the workflow again does not accurately capture CDR flexibility.
We experimentally evaluated ITsFlexible’s predictive performance using three antibodies chosen as challenging test cases. The antibodies had low sequence identity to the training set and the loop length opposed trends observed in the data (long loops for rigid cases and shorter loops for flexible cases). Using cryo-EM, we imaged the antibodies in complex with their antigen and analysed heterogeneity in the density maps50,51. For two of the three cases, the experimental evidence confirmed our predictions. Among the correctly predicted cases, one rigid and one flexible, the former was three residues longer, further demonstrating that the model detects features beyond loop length associated with flexibility. The third antibody featured an even shorter CDRH3 predicted to be flexible, although with lower confidence, and experiments showed no evidence of conformational heterogeneity. Overall, cryo-EM experiments provide strong evidence that ITsFlexible is predictive of real CDR3 conformational dynamics observed in solution.
One limitation of this work is the approach of identifying flexibility. We label a CDR as flexible if we observe multiple conformational states irrespective of whether the antigen receptor is bound to the antigen or in its free state. Previous studies showed that the bound conformation is frequently included in the unbound ensemble, suggesting that conformational selection is a more common mechanism of antibody binding than induced fit4,52,53. We, therefore, expect that ITsFlexible predictions are representative of the flexibility of unbound CDRs. This assumption is supported by the agreement with MD simulations of unbound antigen receptors that we observe. As some functional properties (such as binding affinity) are dependent on the balance of flexibility in bound and unbound states7, we suggest the prediction of changes in CDR flexibility on binding as an important future direction. Owing to the limited amount of data capturing CDR conformational states, however, predicting flexibility conditional on the antigen is currently challenging.
In this work, we present ALL-conformations41 and ITsFlexible (available at https://github.com/oxpig/ITsFlexible and in ref. 42). ALL-conformations captures the full range of experimentally observed conformational diversity of loops between antiparallel β-strands, enabling a detailed analysis of loop dynamics and supporting the training and benchmarking of more robust conformation prediction workflows. ITsFlexible accurately predicts the CDR conformational flexibility and is able to help with key problems in drug design. CDR flexibility has been linked to reduced binding affinity7,8 and increased polyspecificity5,6, both of which are typically undesirable when engineering antigen receptors for therapeutic use10. By allowing the rapid screening of candidate molecules, ITsFlexible can help identify receptors with more favourable therapeutic profiles. Moreover, flexibility predictions can help identify antigen receptors for which computationally expensive MD simulations are likely to yield the greatest benefit and support a more strategic use of computational resources. Several studies have shown that incorporating structural ensembles of flexible molecules can improve downstream tasks, such as antibody–antigen docking54,55. Taken together, the development of ALL-conformations and ITsFlexible paves the way for tackling more complex tasks such as sampling structures of different conformational states in the future.
Don’t miss more hot News like this! Click here to discover the latest in AI news!
2025-10-16 00:00:00