
A multimodal conversational agent for DNA, RNA and protein tasks
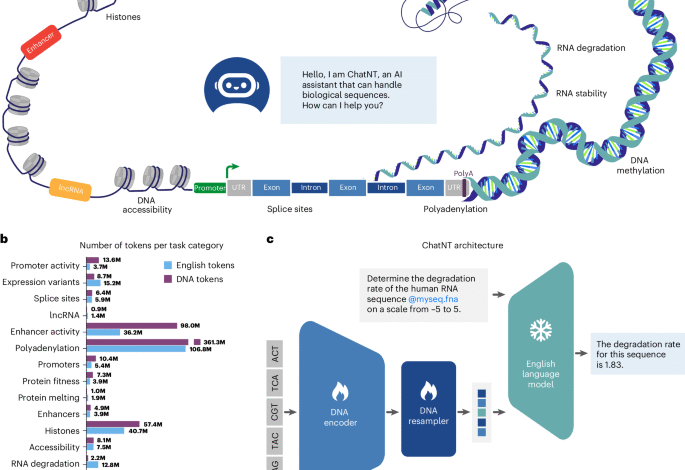
Transforming DNA foundation models into conversational agents with ChatNT
ChatNT is a framework for genomics instruction tuning, extending instruction-tuning agents to the multimodal space of biology and biological sequences. Our framework is designed to be modular and trainable end to end. It combines (1) a DNA encoder model, pretrained on genome sequencing data and that provides DNA sequence representations; (2) an English decoder, typically a pretrained GPT-style LLM, to comprehend the user instructions and produce responses; and (3) a projection layer that projects the representations extracted by the DNA encoder into the embedding space of the input English words, such that both can be used by the English decoder (Fig. 1c and Methods). In contrast to most multimodal works (for example, ref. 26) that would typically freeze the encoder and train only the projection, and sometimes the decoder, we decided in this work to backpropagate the gradients in the encoder in addition to the projection to allow supervised knowledge propagation at the DNA model level. As the English decoder is kept frozen, ChatNT benefits from its entire initial conversational capabilities, ensuring these do not degrade during training. In this work, we use the Nucleotide Transformer v2 (500-million-parameter model pretrained on genomes from 850 species) as the DNA encoder15 and Vicuna-7b (instruction-fine-tuned LLaMA model with 7 billion parameters) as the English decoder32 to build the conversational agent ChatNT. Keeping this modular architecture allows one to use constantly improving encoders and decoders in the future without changing the model architecture.
a, An illustration of the different categories of downstream tasks included during training. UTR, untranslated region. b, Statistics on the number of English and DNA tokens available for each task in our genomics instructions dataset. English question–answer instructions are tokenized with the LLaMA tokenizer30, while DNA sequences are tokenized using the Nucleotide Transformer tokenizer15. c, The ChatNT approach to build a multimodal and multitask genomics AI system. The ChatNT conversational agent can be prompted in English to solve various tasks given an input question and nucleotide sequence. In this example, the user inputs a DNA sequence (fasta file) and asks the agent to evaluate the degradation rate of the given RNA sequence. The question tokens are combined with the projected DNA representations before passing through the English language model decoder. The pretrained decoder writes the answer through next-token prediction, in this case predicting the degradation rate of the input sequence.
To train and evaluate ChatNT, we converted datasets of genomics tasks into instructions datasets by framing each task in English (Supplementary Fig. 1; see Methods and the following ‘Results’ sections). We created for every task a train and test file each containing the respective DNA sequences combined with curated questions and answers in English. See Fig. 1c for an example of question and answer for predicting RNA degradation levels: ‘User: Determine the degradation rate of the human RNA sequence @myseq.fna on a scale from −5 to 5. ChatNT: The degradation rate for this sequence is 1.83.’, where the projected embeddings of the candidate DNA sequence are inserted at the @myseq.fna position. We keep the same train–test splits as the original sources of each task and use different questions for train and test to assess the English generalization capabilities of the model. This allows one to evaluate not only the agent capability to generalize between DNA sequences but also its robustness to the English language used. We also provide a flexible way to interleave English and DNA sequences through the usage of positional tags (@myseq.fna), allowing users to refer to several sequences in the same question.
ChatNT is trained to solve all tasks simultaneously, with a uniform sampling over tasks per batch. Multitasking is achieved by ChatNT by prompting in natural language, where the question asked by the user will guide the agent towards the task of interest. Given a text prompt and one or multiple DNA sequences as input, ChatNT is trained to minimize a unified objective for all tasks, which takes the form of the cross-entropy loss between ChatNT predictions and the target answer tokens, as in other instruction fine-tuning works32,33. This single objective allows one to learn seamlessly across tasks without introducing conflicting gradients or scale issues coming from different objectives and loss functions (for example, cross-entropy for classification versus mean squared error (MSE) for regression). In addition, it allows us to extend the model with additional tasks in the future without requiring changes in the model architecture or training it from scratch. In summary, ChatNT provides a general genomics AI system that solves multiple tasks in a conversational manner, thus providing a different paradigm for genomics models.
In addition to seamlessly integrating multiple types of labelled and experimental data into a single general foundation model, ChatNT is designed to be conversational to enable users to easily interact with it and to use it without requiring a programming background (see examples in Supplementary Fig. 1). We rely on a frozen English language model, Vicuna-7b32, that has been instruction fine-tuned from LLaMA30. ChatNT keeps all the intrinsic conversational capabilities of the language model. Interestingly, we observed that, as the training dataset used to build LLaMA already contained a large set of life sciences papers, our agent is also capable to answer multiple questions about genomics, including defining regulatory elements such as promoters and enhancers, zero shot, that is, without any additional training data. In addition, ChatNT can answer numerous non-biology-related questions and solve tasks such as summarizing or writing simple programming code. As our approach is general and builds on top of any pretrained English language model, ChatNT capabilities can improve organically with new and more powerful open-sourced language models. While the conversational capability is an important aspect of ChatNT but is already provided by the respective language model, we focused in this work on demonstrating that the conversational agent ChatNT can solve a wide range of advanced genomics tasks in English with high accuracy.
ChatNT achieves improved performance on a genomics benchmark
To develop ChatNT and optimize its architecture, we created an instructions version of the Nucleotide Transformer benchmark15 (Methods and Supplementary Table 1). This collection of genomic datasets is suitable for fast iteration during model experimentation as it contains a varied panel of small-sized datasets and has been extensively evaluated in multiple studies of DNA foundation models15,17. We trained ChatNT to solve all 18 tasks at once and in English and evaluated its performance on test set DNA sequences and questions.
We first used this benchmark to systematically compare the performance of ChatNT with two different projection architectures. The classical way of aggregating information from the encoder in previous multimodal models is to use a trainable projection to convert the encoder embeddings into language embedding tokens, which have the same dimensionality of the word embedding space in the language model26,27,34. In ChatNT, we used the Perceiver resampler from Flamingo27 based on gated cross-attention as the projection layer (Supplementary Fig. 2a). Using this projection layer and fine-tuning both the DNA encoder and the projection on all 18 tasks, ChatNT obtained a new state-of-the-art accuracy on this benchmark with an average Matthew’s correlation coefficient (MCC) of 0.71, 2 points above the previous state-of-the-art Nucleotide Transformer v2 (500M) model (Fig. 2a and Supplementary Figs. 2d and 3).

a, The average performance of ChatNT, ChatNT with no English-aware projection and 13 different genomics foundation models across all 18 tasks of the Nucleotide Transformer benchmark15. The bar plots represent mean MCC values ± s.e.m. (n = 18). b, A radar plot depicting the performance of ChatNT in each of the 18 tasks compared with specialized Nucleotide Transformer v2 models fine-tuned individually on each task.
However, similar to all other projection layers26,34,35, the current implementation of the Perceiver resampler generates the same fixed set of embeddings for the encoder tokens independently of the question asked, and therefore it needs to capture in this set of embeddings all relevant information for every downstream task. We hypothesized that this feature can create an information bottleneck in genomics when scaling the model for multiple downstream tasks given the diversity of potential sequences, from different lengths and species, and biological properties. Therefore, we developed an English-aware Perceiver projection that extracts representations from the input sequence dependent on the English question asked by the user, which allows one to leverage contextual information encoded in the input DNA sequences that are relevant for the specific question (Methods and Supplementary Fig. 2b). We observed significantly improved performance by accounting for the question when projecting the DNA embeddings into the English decoder space (average MCC of 0.77 versus 0.71; Fig. 2a and Supplementary Fig. 2c,d). This can be explained by the very context- and task-specific information in DNA sequences that we must retain in order to tackle diverse genomics tasks. Because the decoder remains frozen, the projection layer needs to not only bring the sequence embeddings into the embedding space of the English decoder but also to perform the operations to extract the relevant information from the embedding to answer the question. Our results show that making the projection aware of the question facilitates both aspects, thus achieving a better performance and transfer across tasks.
In summary, ChatNT with an English-aware projection (from now on called just ChatNT) achieves a new state-of-the-art accuracy on this benchmark (average MCC of 0.77) in addition to solving all 18 tasks at once (Fig. 2a). ChatNT improves the average performance by 8 points over the previous state-of-the-art Nucleotide Transformer v2 (500M) model, which was used as the DNA encoder within ChatNT (average MCC of 0.77 versus 0.69; Fig. 2a,b). In addition to generalizing across DNA sequences and tasks, ChatNT also generalizes across questions, as demonstrated by the low variability in the accuracy of the predictions based on the variability of the language prompt (Supplementary Fig. 4). Our results demonstrate that a single unified objective formulated in natural language triggers transfer learning between multiple downstream tasks and helps to deliver improved performance.
A curated instructions dataset of biologically relevant tasks
Although the Nucleotide Transformer benchmark15 was very suitable for model experimentation and to debug the system, it misses many tasks of great biological relevance in genomics related to more complex biological processes as well as more recent experimental techniques and tasks that involve quantitative predictions. Therefore we curated a second genomics instructions dataset containing 27 genomics tasks framed in English derived from different studies that cover several regulatory processes (Methods and Supplementary Table 2). These include tasks related to DNA (21 tasks), RNA (3) and protein sequences (3) from multiple species framed as both binary/multilabel classification and regression tasks. The final instructions dataset contains a total of 605 million DNA tokens, that is, 3.6 billion base pairs, and 273 million English tokens (including an average of 1,000 question–answer pairs per task) (Fig. 1b).
This collection includes a non-redundant subset of tasks from the Nucleotide Transformer15 and the BEND36 benchmarks, complemented with relevant tasks from the plant AgroNT benchmark37 and human ChromTransfer38. These benchmarks have been extensively used in the literature, come from different research groups and represent diverse DNA processes and species. These selected tasks include binary and multilabel classification tasks covering biological processes related to histone and chromatin features, promoter and enhancer regulatory elements, and splicing sites.
We further added state-of-the-art and challenging regression tasks related to promoter activity37, enhancer activity7, RNA polyadenylation11 and degradation10, and multiple protein properties39. These are reference datasets in the respective fields and are related to very complex properties of biological DNA, RNA and protein sequences. All RNA and protein tasks are predicted from the corresponding DNA and coding sequences (CDS) instead of the RNA and protein sequences, respectively. Getting the matching DNA sequence is trivial for RNA sequences but more challenging for protein sequences owing to the complexity of codon usage. Therefore, we used the CDS annotations for protein tasks curated by Boshar et al.39.
See Fig. 3 and Supplementary Fig. 1 for examples of questions and answers for different types of genomics tasks used in our dataset (see also Supplementary Figs. 5–7). For instance, a training example for an enhancer classification task would be ‘User: Is there an enhancer from human cells present in this sequence @myseq.fna, and can you characterize as weak or strong? ChatNT: Yes, a weak enhancer is present within the DNA sequence that you provided.’, where the projected embeddings of the candidate DNA sequence are inserted at the @myseq.fna position. Regression tasks are also framed in English, and the agent needs to write the digits corresponding to the requested quantity: for example, ‘User: Determine the degradation rate of the mouse RNA sequence @myseq.fna on a scale from −5 to 5. ChatNT: The measured degradation rate for this sequence is 2.4.’ (see Methods for details on the quantitative scale). The loss is equally computed as the cross-entropy loss between the predicted and the target answer tokens, treating scalar values as digit tokens. This approach worked well in our setting and yielded results comparable to traditional MSE loss because ChatNT is autoregressive, allowing it to capture the sequential structure of numbers. Errors in earlier digits, such as the order of magnitude, result in higher loss, prompting the model to focus on predicting the most significant digits first before refining the less significant ones, thus effectively introducing a hierarchical decomposition of numbers. For performance evaluation, we extract the digits from each answer and test their correlation with the ground-truth values.

a,d,e, Left: an example of a conversation for promoter (a), DNA methylation (d) and splice sites tasks (e). Right: a heatmap displaying the confusion matrix comparing the predicted labels of ChatNT and observed labels. The performance metric is reported. b,c,f, Left: an example of a conversation for promoter strength in tobacco leaf (b), RNA degradation (c) and protein meltome tasks (f). Right: a scatter plot comparing the predictions of ChatNT and observed values. The PCC is reported.
In summary, this curated set of tasks provides a general perspective of the capabilities and usefulness of our model in different biological sequence domains. We train ChatNT as a general agent to solve all 27 genomics tasks at once and in English, and compare its performance with the state-of-the-art specialized model for each task (Methods).
Evaluation on tasks from various genomics processes and species
We first evaluated the performance of ChatNT on the 21 tasks related to different DNA processes from yeast, plants, fly, mouse and human. ChatNT is competitive with the performance of the different specialized models that were fine-tuned directly on each of these individual tasks, being also robust to the variability of the language prompt (Figs. 3a,b,d,e and 4a,c). In particular, we obtained an improved performance on the detection of human enhancer types. Still, we observed significantly reduced performance for enhancers from plant species when compared with the state-of-the-art AgroNT model fine-tuned specifically on this task37. As AgroNT was pretrained on genomes from 48 diverse plant species, improving the encoder used in ChatNT might lead to improved performance on this type of task.

a, Bar plots with the performance of ChatNT compared with respective baselines per task. The metric used for each task is the same used in the respective baseline study (Supplementary Table 2). Data are presented as mean ± s.d. (n > 10 per task). Chrom., chromatin; Dev., developmental, Hk., housekeeping. b–d, A comparison between ChatNT and baselines for all tasks (n = 27) (b), classification tasks (n = 17) (c) and regression tasks (n = 10) (d). Metrics are the same as in a. The box plots mark the median, upper and lower quartiles and 1.5× interquartile range (whiskers); outliers are shown individually.
As ChatNT solves the tasks in English, it can seamlessly handle binary and multilabel classification tasks. By extracting the term predicted by ChatNT in the answer, we can quantify its predictive performance. As we show for some examples in Fig. 3, ChatNT accurately identifies input sequences with human or mouse promoters (Fig. 3a), with CpG sites methylated in human embryonic stem cells (HUES64 cell line; Fig. 3d) and with splice acceptor and donor sites (Fig. 3e).
ChatNT is also able to solve quantitative tasks by writing the digits of the predicted score. We observed competitive performance on predicting promoter activity in plants, namely tobacco leaves (Fig. 3b) and maize protoplasts, but significantly reduced performance on Drosophila enhancer activity over the state-of-the-art DeepSTARR model7 (Fig. 4a). Importantly, the distributions of the predicted digits correlate well with the original scores (Fig. 3b). This capability to proficiently address regression tasks is of paramount importance in biology and is particularly significant in light of the acknowledged limitations and unreliability of numerical processing in language models40,41. Still, we observed a reduced average performance on regression tasks over classification ones, probably due to the difference in complexity and classification tasks being more represented in the training set. We assume that this might be solved by improving the balance between classification and regression tasks during training, through either a weight loss or a task sampling frequencies curriculum42.
ChatNT solves transcriptomics and proteomics tasks
ChatNT is built with a flexible architecture that allows it to handle any type of biological sequence that can be processed with our DNA encoder, the Nucleotide Transformer15. To showcase its generalization, we have included in the new genomics instructions dataset three RNA and three protein regression tasks (Supplementary Figs. 6 and 7). These include predicting RNA polyadenylation and degradation rates as well as different protein features. Examples of conversations used for model training are: ‘User: What is the measured polyadenylation ratio of the proximal site of the RNA sequence @myseq.fna in human HEK293 cells, considering a range from 0 to 1? ChatNT: That sequence has a polyadenylation ratio of the proximal site of 0.69.’ and ‘User: Specify the melting point of the protein with the given coding sequence (CDS) @myseq.fna within the 0 to 100 range. ChatNT: This protein demonstrates a melting point of 80.81.’. The performance of ChatNT was compared with the state-of-the-art specialized models APARENT2 for polyadenylation11, Saluki for RNA degradation10 and ESM2 for the protein tasks31 (Supplementary Table 2).
Overall, we observed good performance for ChatNT on the test sets of the six RNA and protein tasks, with Pearson correlation coefficients (PCCs) between 0.62 and 0.91 (Figs. 3c,f and 4a). ChatNT outperformed the specialized models for the prediction of proximal polyadenylation site ratio (PCC of 0.91 versus 0.90) and protein melting points (PCC of 0.89 versus 0.85). Regarding the RNA degradation tasks in human and mouse, ChatNT obtained a PCC of 0.62 and 0.63, 10 points below the specialized Saluki model10 (PCC of 0.74 and 0.71). ChatNT also obtained competitive performance with the state-of-the-art protein language model ESM231 on the two other protein tasks related to protein fluorescence and stability. Although ChatNT cannot yet outperform every specialized model on RNA and protein tasks, we show that it can already handle such tasks and achieve high performance using the DNA foundation model Nucleotide Transformer as a DNA encoder. ChatNT’s flexible architecture allows one to plug in different encoders, such as language models specialized for RNA43,44 and protein domains31, which should reduce the gap to specialized deep learning models in the transcriptomics and proteomics fields and improve the capabilities and generalization of ChatNT towards a unified model of biology.
Assessing the confidence of ChatNT answers
ChatNT is built to assist and augment scientists and researchers in their daily research. As such, its performance and reliability are paramount. However, in contrast to standard machine learning models that return probabilities or quantitative scores, ChatNT directly answers questions, preventing the user from getting a sense of its confidence and, thus, reducing its practical value for sensitive applications. This is an important challenge and common to all current conversational agents24,26. To address this, we investigated a way to assess the confidence of our agent for binary classification tasks. Instead of directly generating answers to the binary classification question for a given sequence, we compute the model perplexity for that question over examples of both positive and negative answers. We make sure that these selected answers were not included in the model training dataset. Those perplexity values towards positive and negative answers are then used to derive logits and probabilities for each class for the candidate question. This method allows us to derive probabilities from ChatNT for each question example, similar to standard classifiers, and we refer to it as the perplexity-based classifier (Fig. 5a). Currently, this process is studied as a post-hoc analysis, not yet integrated into the ChatNT tool, but it could be incorporated into future versions once fully developed to provide this information to the user.

a, A cartoon describing the perplexity-based classifier based on ChatNT answers. b, A calibration plot for the task of human chromatin (chrom.) accessibility (cell line HepG2). The scatter plot compares the predicted probability and fraction of positives over ten bins for the original (green) and calibrated (purple) perplexity-based classifiers. c, A histogram with the predicted probability over ten bins for the original perplexity-based classifiers. d, A histogram with the predicted probability over ten bins for the calibrated perplexity-based classifiers. e, A comparison of the performance (MCC) of the ChatNT answers (yes versus no) and its derived perplexity-based probabilities for all binary classification tasks.
Computing probabilities enables us to assess the calibration of the model, that is, the correlation between the predicted probability, its confidence and the accuracy of its prediction. We say that a model is well calibrated when a prediction of a class with confidence p is correct 100% of the time. We computed the ChatNT perplexity-based probabilities for all binary classification tasks. In Fig. 5b–d, we show an example of a calibration plot based on the predictions for the chromatin accessibility task. We observe that our model is well calibrated for low- and high-confidence areas, but less in medium-confidence ones. For instance, examples predicted with a probability of 0.9 are correctly predicted 90% of the time, while examples predicted with probability 0.5 are correctly predicted only 25% of the time. To improve this, we show that we can calibrate our model by fitting on the training set a Platt’s model45, to improve the confidence of the model across all ranges of predictions (Fig. 5b–d). This calibration step is performed for all binary classification tasks. Overall, we achieve the same performance for ChatNT across tasks using these perplexity-based predictions (Fig. 5e) but with improved calibration. As a consequence, our approach can accurately measure the predictive performance of a language model in addition to effectively assessing its uncertainty level. This technique, while being general, should also be beneficial to other language model fields.
Model interpretation reveals learned DNA sequence features
A key application of language models for biological sequences has been to uncover the underlying code or grammar of DNA, RNA and protein sequences. To evaluate whether ChatNT’s predictions rely on biologically relevant sequence features and could be used for further discoveries, we used model interpretation tools across hundreds of sequences from different tasks in our dataset. While these techniques have been widely applied for sequence models, notably in the case of DNA7,46, this has never been done for multimodal models where the output is expressed in natural language such as ChatNT. To do that, we quantified the contribution of each DNA token, that is, 6-mer, within the input DNA sequence to the predicted English tokens answered by ChatNT (Fig. 6a). More specifically, we calculated the gradient of the predicted token with respect to the input DNA tokens47, backpropagating through the English decoder, the perceiver projection and the DNA encoder. We applied this approach to sequences from three different genomics tasks for which clear predictive sequence features are known in the literature: splice donor sites that usually overlap a GT dinucleotide, splice acceptor sites that overlap a AG dinucleotide and promoters that rely on the TATA-box motif.

a, A cartoon describing the interpretation of ChatNT predictions with regard to the input sequence. For a given input question and DNA sequence, we compute the gradients of each predicted English token towards every input DNA token. w.r.t., with respect to. b–d, Sequence features extracted from ChatNT for the tasks splice donors (b), splice acceptors (c) and TATA promoter (d). Shown are the consolidated sequence motifs with the y axis as bits (top) and the k-mer spectrum across sequences per task (bottom).
For each task, we computed the contribution of input DNA tokens with respect to the ‘yes’ or ‘no’ token answered by ChatNT. We performed this analysis across a subset of test set sequences per label per task, identifying the highest attributed DNA tokens per input sequence. Displaying the frequency of important 6-mers across all sequences revealed that positively labelled sequences were enriched for known sequence features such as the splice donor and acceptor dinucleotides (Fig. 6b–d). Indeed, combining the top-scoring tokens across all positive sequences and calculating the nucleotide frequencies per position recovers the main sequence pattern per task that matches the known splice donor GT dinucleotide, splice acceptor AG dinucleotides and promoter TATA motif. Altogether, these results demonstrate that ChatNT’s answers are based on biologically coherent features and that the model can be used to interpret the underlying language of DNA. It is important to emphasize that ChatNT accomplishes this with a single, unified model. Typically, such analysis usually relies on specialized or fine-tuned models tailored to individual tasks. The fact that our results reveal consistent and meaningful features extracted across diverse tasks underscores the capacity of ChatNT to learn a generalized grammar of DNA and to connect it to natural language.
Don’t miss more hot News like this! Click here to discover the latest in AI news!
2025-06-06 00:00:00



