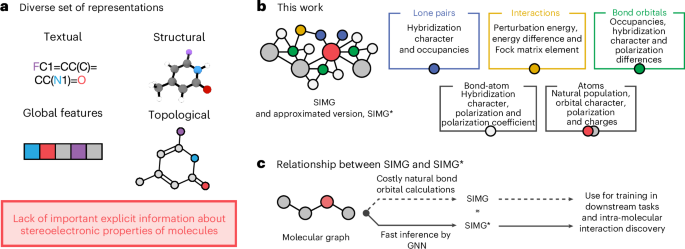
Advancing molecular machine learning representations with stereoelectronics-infused molecular graphs
Hoffmann, R. & Laszlo, P. Representation in chemistry. Angew. Chem. Int. Ed. Engl. 30, 1–16 (1991).
Google Scholar
Cooke, H. A historical study of structures for communication of organic chemistry information prior to 1950. Org. Biomol. Chem. 2, 3179 (2004).
Google Scholar
Springer, M. T. Improving students’ understanding of molecular structure through broad-based use of computer models in the undergraduate organic chemistry lecture. J. Chem. Educ. 91, 1162–1168 (2014).
Google Scholar
Gómez-Bombarelli, R. et al. Design of efficient molecular organic light-emitting diodes by a high-throughput virtual screening and experimental approach. Nat. Mater. 15, 1120–1127 (2016).
Google Scholar
Zhavoronkov, A. et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 37, 1038–1040 (2019).
Google Scholar
Dara, S., Dhamercherla, S., Jadav, S. S., Babu, C. M. & Ahsan, M. J. machine learning in drug discovery: a review. Artif. Intell. Rev. 55, 1947–1999 (2022).
Google Scholar
Gallegos, L. C., Luchini, G., St. John, P. C., Kim, S. & Paton, R. S. Importance of engineered and learned molecular representations in predicting organic reactivity, selectivity, and chemical properties. Acc. Chem. Res. 54, 827–836 (2021).
Google Scholar
Sandfort, F., Strieth-Kalthoff, F., Kühnemund, M., Beecks, C. & Glorius, F. A structure-based platform for predicting chemical reactivity. Chem 6, 1379–1390 (2020).
Google Scholar
Ross, J. et al. Large-scale chemical language representations capture molecular structure and properties. Nat. Mach. Intell. 4, 1256–1264 (2022).
Google Scholar
Yang, Z., Chakraborty, M. & White, A. D. Predicting chemical shifts with graph neural networks. Chem. Sci. 12, 10802–10809 (2021).
Google Scholar
Zhou, J. et al. Graph neural networks: a review of methods and applications. AI Open 1, 57–81 (2020).
Google Scholar
Fang, X. et al. Geometry-enhanced molecular representation learning for property prediction. Nat. Mach. Intell. 4, 127–134 (2022).
Google Scholar
Batzner, S. et al. E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials. Nat. Commun. 13, 2453 (2022).
Google Scholar
Qi, Y., Gong, W. & Yan, Q. Bridging deep learning force fields and electronic structures with a physics-informed approach. Preprint at https://doi.org/10.48550/arXiv.2403.13675 (2024).
Fabrizio, A., Briling, K. R. & Corminboeuf, C. SPAHM: the spectrum of approximated Hamiltonian matrices representations. Digital Discovery 1, 286–294 (2022).
Google Scholar
Elton, D. C., Boukouvalas, Z., Butrico, M. S., Fuge, M. D. & Chung, P. W. Applying machine learning techniques to predict the properties of energetic materials. Sci. Rep. 8, 9059 (2018).
Google Scholar
Rupp, M., Tkatchenko, A., Müller, K.-R. & von Lilienfeld, O. A. Fast and accurate modeling of molecular atomization energies with machine learning. Phys. Rev. Lett. 108, 058301 (2012).
Google Scholar
Pozdnyakov, S. N. & Ceriotti, M. Smooth, exact rotational symmetrization for deep learning on point clouds. Preprint at https://doi.org/10.48550/arXiv.2305.19302 (2023).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Google Scholar
Černý, J. & Hobza, P. Non-covalent interactions in biomacromolecules. Phys. Chem. Chem. Phys. 9, 5291 (2007).
Google Scholar
Anighoro, A. in Quantum Mechanics in Drug Discovery (ed. Heifetz, A.) 75–86 (Humana, Springer, 2020).
Wheeler, S. E., Seguin, T. J., Guan, Y. & Doney, A. C. Noncovalent interactions in organocatalysis and the prospect of computational catalyst design. Acc. Chem. Res. 49, 1061–1069 (2016).
Google Scholar
Weinhold, F. & Landis, C. R. Natural bond orbitals and extensions of localized bonding concepts. Chem. Educ. Res. Pract. 2, 91–104 (2001).
Google Scholar
Llenga, S. & Gryn’ova, G. Matrix of orthogonalized atomic orbital coefficients representation for radicals and ions. J. Chem. Phys. 158, 214116 (2023).
Google Scholar
Bartók, A. P., Kondor, R. & Csányi, G. On representing chemical environments. Phys. Rev. B 87, 184115 (2013).
Google Scholar
Prokhorenkova, L., Gusev, G., Vorobev, A., Dorogush, A. V. & Gulin, A. Catboost: unbiased boosting with categorical features. Adv. Neur. Inf. Proc. Syst. 31, 6638–6648 (2018).
NVIDIA. MegaMolBART. GitHub https://github.com/NVIDIA/MegaMolBART (2022).
Heid, E. et al. Chemprop: a machine learning package for chemical property prediction. J. Chem. Inf. Model 64, 9–17 (2024).
Google Scholar
Alabugin, I. V. Stereoelectronic Effects: A Bridge Between Structure and Reactivity (Wiley, 2016).
Echenique, P. & Alonso, J. L. A mathematical and computational review of Hartree–Fock SCF methods in quantum chemistry. Mol. Phys. 105, 3057–3098 (2007).
Google Scholar
Burke, K. & Wagner, L. O. DFT in a nutshell. Int. J. Quantum. Chem. 113, 96–101 (2013).
Google Scholar
Goerigk, L. & Grimme, S. Double-hybrid density functionals. Wiley Interdiscip. Rev. Comput. Mol. Sci. 4, 576–600 (2014).
Google Scholar
Kneiding, H. et al. Deep learning metal complex properties with natural quantum graphs. Digital Discovery 2, 618–633 (2023).
Google Scholar
Johnson, E. R. et al. Revealing noncovalent interactions. J. Am. Chem. Soc. 132, 6498–6506 (2010).
Google Scholar
Axelrod, S. & Gómez-Bombarelli, R. GEOM, energy-annotated molecular conformations for property prediction and molecular generation. Sci. Data 9, 185 (2022).
Google Scholar
Malinin, A., Prokhorenkova, L. & Ustimenko, A. Uncertainty in gradient boosting via ensembles. Preprint at https://doi.org/10.48550/arXiv.2006.10562 (2020).
Chua, K., Calandra, R., McAllister, R. & Levine, S. Deep reinforcement learning in a handful of trials using probabilistic dynamics models. In Proc. 32nd International Conference on Neural Information Processing Systems 4759–4770 (NIPS, 2018).
Goan, E. & Fookes, C. in Case Studies in Applied Bayesian Data Science (eds Mengerson, K. L. et al.) 45–87 (Springer, 2020).
Beluch, W. H., Genewein, T., Nurnberger, A. & Kohler, J. M. The power of ensembles for active learning in image classification. In Proc. 2018 IEEE/CVF Conference on Computer Vision and Pattern Recognition 9368–9377 (IEEE, 2018).
León, I., Alonso, E. R., Cabezas, C., Mata, S. & Alonso, J. L. Unveiling the n→π* interactions in dipeptides. Commun Chem 2, 3 (2019).
Google Scholar
Newberry, R. W., Bartlett, G. J., VanVeller, B., Woolfson, D. N. & Raines, R. T. Signatures of n→π* interactions in proteins. Protein Sci. 23, 284–288 (2014).
Google Scholar
Hodges, J. A. & Raines, R. T. Energetics of an n → π* interaction that impacts protein structure. Org Lett 8, 4695–4697 (2006).
Google Scholar
Zhou, Y., Morais-Cabral, J. H., Kaufman, A. & MacKinnon, R. Chemistry of ion coordination and hydration revealed by a K+ channel–Fab complex at 2.0 Å resolution. Nature 414, 43–48 (2001).
Google Scholar
Bartlett, G. J., Choudhary, A., Raines, R. T. & Woolfson, D. N. n→π* interactions in proteins. Nat. Chem. Biol. 6, 615–620 (2010).
Google Scholar
dos Passos Gomes, G. & Alabugin, I. V. Drawing catalytic power from charge separation: stereoelectronic and zwitterionic assistance in the Au(I)-catalyzed Bergman cyclization. J. Am. Chem. Soc. 139, 3406–3416 (2017).
Google Scholar
Gomes, G. D. P., Vil’, V., Terent’ev, A. & Alabugin, I. V. Stereoelectronic source of the anomalous stability of bis-peroxides. Chem. Sci. 6, 6783–6791 (2015).
Google Scholar
Grabowski, S. J. Tetrel bond–σ-hole bond as a preliminary stage of the SN2 reaction. Phys. Chem. Chem. Phys. 16, 1824–1834 (2014).
Google Scholar
Sarazin, Y., Liu, B., Roisnel, T., Maron, L. & Carpentier, J.-F. Discrete, solvent-free alkaline-earth metal cations: metal···fluorine interactions and ROP catalytic activity. J. Am. Chem. Soc. 133, 9069–9087 (2011).
Google Scholar
Ramakrishnan, R., Dral, P. O., Rupp, M. & von Lilienfeld, O. A. Quantum chemistry structures and properties of 134 kilo molecules. Sci. Data 1, 140022 (2014).
Google Scholar
Mardirossian, N. & Head-Gordon, M. ωB97M-V: a combinatorially optimized, range-separated hybrid, meta-GGA density functional with VV10 nonlocal correlation. J. Chem. Phys. 144, 214110 (2016).
Google Scholar
Shao, Y. et al. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys. 113, 184–215 (2015).
Google Scholar
Glendening, E. D., Landis, C. R. & Weinhold, F. NBO 7.0: new vistas in localized and delocalized chemical bonding theory. J. Comput. Chem. https://doi.org/10.1002/jcc.25873 (2019).
Ong, S. P. et al. Python Materials Genomics (pymatgen): a robust, open-source python library for materials analysis. Comput. Mater. Sci. 68, 314–319 (2013).
Google Scholar
Blau, S., Spotte-Smith, E. W. C., Wood, B., Dwaraknath, S. & Persson, K. Accurate, automated density functional theory for complex molecules using on-the-fly error correction. Preprint at chemRxiv https://doi.org/10.26434/chemrxiv.13076030.v1 (2020).
Mathew, K. et al. Atomate: A high-level interface to generate, execute, and analyze computational materials science workflows. Comput. Mater. Sci. 139, 140–152 (2017).
Google Scholar
Paszke, A. et al. PyTorch: an imperative style, high-performance deep learning library. In Advances in Neural Information Processing Systems Vol. 32 (eds. Wallach, H. et al.) 8024–8035 (Curran Associates, Inc., 2019).
Falcon, W. A. et al. PyTorch Lightning. GitHub https://github.com/PyTorchLightning/pytorch-lightning (2019).
Fey, M. & Lenssen, J. E. Fast graph representation learning with PyTorch Geometric. Preprint at https://doi.org/10.48550/arXiv.1903.02428 (2019).
Corso, G., Cavalleri, L., Beaini, D., Liò, P. & Veličković, P. Principal neighbourhood aggregation for graph nets. Preprint at https://doi.org/10.48550/arXiv.2004.05718 (2020).
Li, G., Müller, M., Thabet, A. & Ghanem, B. DeepGCNs: can GCNs go as deep as CNNs? Preprint at https://doi.org/10.48550/arXiv.1904.03751 (2019).
Godwin, J. et al. Simple GNN regularisation for 3D molecular property prediction & beyond. Preprint at https://doi.org/10.48550/arXiv.2106.07971 (2021).
Veličković, P. et al. Graph attention networks. Preprint at https://doi.org/10.48550/arXiv.1710.10903 (2017).
Cai, C. & Wang, Y. A note on over-smoothing for graph neural networks. Preprint at https://doi.org/10.48550/arXiv.2006.13318 (2020).
Boiko, D. et al. Advancing molecular machine learned representations with stereoelectronics-infused molecular graphs. Zenodo https://doi.org/10.5281/zenodo.14393496 (2024).
Don’t miss more hot News like this! Click here to discover the latest in AI news!
2025-05-23 00:00:00