Generalized biological foundation model with unified nucleic acid and protein language
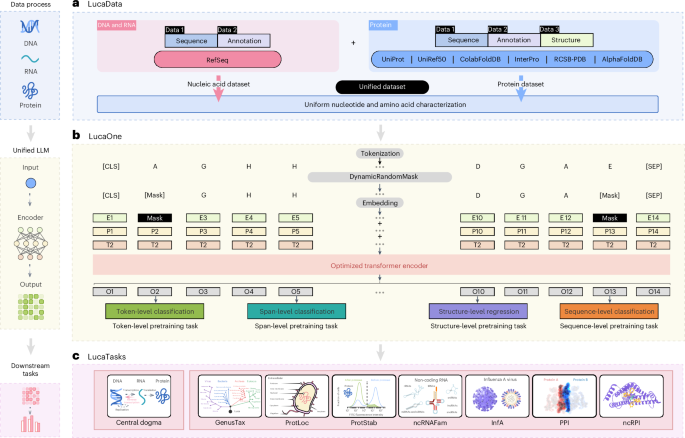
Model architecture
Figure 1b illustrates the design of LucaOne, which utilizes the transformer-encoder21 architecture with the following enhancements:
-
(1)
The vocabulary of LucaOne comprises 39 tokens, including both nucleotide and amino acid symbols (refer to ‘Vocabulary’).
-
(2)
The model employs pre-layer normalization over post-layer normalization, facilitating the training of deeper networks45.
-
(3)
Rotary position embedding46 is implemented instead of absolute positional encoding, enabling the model to handle sequences longer than those seen during training.
-
(4)
It incorporates mixed training of nucleic acid and protein sequences by introducing token-type embeddings, assigning 0 for nucleotides and 1 for amino acids.
-
(5)
Besides the pre-training masking tasks for nucleic acid and protein sequences, eight semi-supervised pre-training tasks have been implemented based on selected annotation information (refer to ‘Pre-training tasks details’).
Vocabulary
The vocabulary of LucaOne consists of 39 tokens. Owing to the unified training of nucleic acid and protein sequences, the vocabulary includes 4 nucleotides (‘A’, ‘T’, ‘C’ and ‘G’) of nucleic acid (‘U’ compiled ‘T’ in RNA), ‘N’ for unknown nucleotides, 20 amino acids of protein (20 uppercase letters excluding ‘B’, ‘J’, ‘O’,‘U’, ‘X’ and ‘Z’), ‘X’ for unknown amino acids, ‘O’ for pyrrolysine, ‘U’ for selenocysteine, other 3 letters (‘B’, ‘J’ and ‘Z’) not used by amino acids, 5 special tokens (‘[PAD]’, ‘[UNK]’,‘[CLS]’,‘[SEP]’ and ‘[MASK]’), and 3 other ‘.’, ‘-’ and ‘*’). Owing to the amino acid letters overlapping with the nucleotide letters, the use of ‘1’, ‘2’, ‘3’, ‘4’ and ‘5’ instead of ‘A’, ‘T’, ‘C’, ‘G’ and ‘N’, respectively.
Pre-training data details
Nucleic acid
The nucleic acid was collected from the NCBI RefSeq genome database, involving 297,780 assembly accessions. The molecular types included DNA and RNA (Fig. 2a). The DNA sequence, DNA selected annotation, RNA sequence and RNA selected annotation were obtained from the format files ’genomic.fna’, ’genomic.gbff’, ’rna.gbff’ and ’rna.fna’, respectively. Among all pre-training sequences, 70% of DNA sequences and 100% of RNA sequences were derived from annotated genomes, while the remaining unannotated sequences were retained to ensure diversity.
DNA reverse strand: the DNA dataset expanded reverse strand sequences with their annotation. A total of 23,095,687 reverse-strand DNA sequences were included.
Genome region types: eight important genome region types in nucleic acids were selected, including ‘CDS’, ‘intron’, ‘tRNA’, ‘ncRNA’, ‘rRNA’, ‘miscRNA’, ‘tmRNA’ and ‘regulatory’. Each nucleotide in the sequence had a label index of 8 categories (0–7) or −100 when it did not belong to these 8 categories.
Order-level taxonomy: the order-level label of the taxonomy tree was selected as the classification label of the nucleic acid sequence. Each sequence had a label index of 735 categories (0–734) or −100 without the order-level taxonomy.
Segmentation: owing to the limited computing resources, each nucleic acid sequence was segmented according to a given maximum length. The fragmentation strategy was presented in Supplementary Fig. 2.
Protein
Protein sequence data were obtained from UniRef50, UniProt and ColabFoldDB. UniRef50 was added to the UniProt database to sample high-quality representative sequences, while ColabFoldDB was incorporated to enhance the diversity of protein sequences. For ColabFoldDB, redundancy within each cluster was minimized by retaining only the ten most diverse sequences. Duplicated sequences between UniProt and ColabFoldDB were excluded. Sequence, taxonomy and keywords were collected from UniProt and ColabFoldDB. The sites, domains and homology regions were extracted from Interpro. The tertiary structure was derived from RCSB-PDB and AlphaFold2-Swiss-Prot.
Sequence: the right truncation strategy was applied when the sequence exceeded the maximum length.
Order-level taxonomy: order-level classification information is used as the protein sequence taxonomy. There were 2,196 categories; each sequence had a label index (0–2,195) or −100 if its order-level information was missing.
Site: four types of site regions (‘active site’, ‘binding site’, ‘conserved site’ and ‘PTM’) with 946 categories were included. For each amino acid in a sequence, if it was a site location, there was a label index (0–945); otherwise, it was marked with −100.
Homology: a homologous superfamily is a group of proteins that share a common evolutionary origin with a sequence region, reflected by similarity in their structure. There were 3,442 homologous region types; each amino acid in these regions had a label index (0–3,441) corresponding to its type, and the other amino acids were labelled −100.
Domain: domain regions are distinct functional, structural or sequence units that may exist in various biological contexts. A total of 13,717 domain categories were included; each amino acid in these regions had a label index (0–13,716) corresponding to its category, and the other amino acids were marked with −100.
Keyword: keywords are generated based on functional, structural or other protein categories. Each sequence was labelled as a set of label indices with 1,179 keywords or −100 without keywords.
Structure: the spatial coordinates of the Cα atom were used here as the amino acid coordinates. Each amino acid was labelled with a three-dimensional coordinate normalized within the protein chain (preferentially selected the structure from RCSB-PDB). The amino acids at the missing locations were labelled (−100, −100, −100). In total, only about half a million protein sequences had structural information.
Pre-training tasks details
LucaOne has employed a semi-supervised learning approach to enhance its applicability in biological language modelling. Bioinformatics analysis often involves different modalities for input and output data, and most downstream tasks extend from understanding nucleic acid or protein sequences, so our pre-training tasks have been augmented with eight foundational sequence-based annotation categories. These annotations complement the self-supervised masking task, facilitating more effective learning for improved performance in downstream applications. The selection criteria for these annotations focused on universality, lightweight design and high confidence level; consequently, only a subset of the data has such annotations. As listed in Supplementary Fig. 3, there are ten specific pre-training tasks at four levels. All loss functions are presented in Supplementary Note 1.
Token-level tasks: Gene-Mask and Prot-Mask tasks randomly mask nucleotides or amino acids in the sequence following the BERT masking scheme47 and predict these masked nucleotides or amino acids based on the sequence context in training.
Span-level tasks: the model is trained to recognize some essential regions based on the sequence context. For nucleic acid sequences, eight essential genome region types are learned. For protein sequences, three types of region are labelled: site, homology and domain regions.
Sequence-level tasks: Gene-Taxonomy, Prot-Taxonomy and Prot-Keyword are the order-level taxonomies of nucleic acid, protein and protein-tagged keywords, respectively. They are all sequence-level learning tasks.
Structure-level tasks: as the structure of a protein determines its function, we use a small amount of protein data with a tertiary structure for simple learning in the pre-training phase. Instead of learning the spatial position at the atomic level, the spatial position of amino acids is trained (using the position of the Cα atom as the position of the amino acid).
Pre-training information
On the dimensions of the embedding, the research conducted by Elnaggar et al.11 demonstrates that the ESM2-3B (embedding dimension 2,560) model outperforms the 650 million (embedding dimension 1,280) version, while the 15 billion (embedding dimension: 5,120) version does not consistently improve performance and substantially increases the computational burden. For the relationship between model size and training data size, Hoffmann et al. suggest that a minimum of 20.2 billion tokens is essential to adequately train a 1 B-sized model48.
The critical hyperparameters we adopted are as follows: the architecture of LucaOne consists of 20 transformer-encoder blocks with 40 attention heads each, supports a maximal sequence length of 1,280 and features an embedding dimension of 2,560. The model is composed of a total of 1.8 billion parameters. We employed 10 different pretraining tasks, assigning an equal weight of 1.0 to the Gene-Mask, Prot-Mask, Prot-Keyword and Prot-Structure tasks, while assigning a reduced weight of 0.2 to the remaining tasks to equilibrate task complexity (Supplementary Note 1, equation (11)). We used the AdamW optimizer with betas (0.9, 0.98) and a maximum learning rate of 2 × 10−4, incorporating a linear warm-up schedule throughout the learning-rate updates. For the model training regimen, we utilized a batch size of 8 coupled with a gradient accumulation step of 32. The model underwent training on 8 Nvidia A100 graphics processing units spanning 120 days. A model checkpoint of 5.6 million (5.6 million, trained with 36.95 billion tokens) was selected for the subsequent validation tasks, aligned with ESM2-3B in terms of the volume of data trained for comparison.
To elucidate the advantages of mixed training involving both nucleic acids and proteins, we further conducted experiments with two supplementary models, LucaOne-Gene and LucaOne-Prot, trained exclusively with nucleic acids and proteins, respectively. Their performance in the central dogma of the biology task was evaluated with the same checkpoint (5.6 million) of the two models.
Checkpoint selection criteria: we have released the 5.6 million checkpoint aligned with the ESM2-3B model for a comparable volume of data trained, which was trained with 36.95 billion tokens over 20 times the model’s parameters. We also released the 17.6 million checkpoint (trained with 116.62 billion tokens) based on three criteria: (1) the loss curve slowly descended after 17.6 million steps during training (Extended Data Fig. 3a); (2) the losses are relatively stable on the validation and testing set between 15 million and 20 million steps, making 17.6 million optimal (Extended Data Fig. 3b,c); (3) the improvement in the performance of representative downstream tasks is very limited. For example, in the ncRPI task, the accuracy is 94.93% at checkpoint 17.6 million, which is only a marginal improvement compared to an accuracy of 94.78% at checkpoint 5.6 million (Extended Data Fig. 3d).
LucaOne embeddings-level analysis
Details of t-SNE datasets: the S1 dataset was curated from marine data available in CAMI249, selecting contigs with lengths ranging from 300 to 1,500 nucleotides. The contigs of each species were redundant by MMSeqs, employing a coverage threshold of 80% and sequence identity of 95%, culminating in a collection of 37,895 nucleic acid contigs from 12 species. We randomly selected 100 samples from each species, totalling 1,200 items for visualization.
The S2 dataset originated from clan data within Pfam, maintaining clan categories with a minimum of 100 Pfam entries, resulting in 189,881 protein sequences across 12 clan categories. For visualization, we randomly selected one sample for each Pfam entry in every clan, amounting to 2,738 samples.
The S3 dataset was selected from the UniProt database from 1 May 2023 to 16 December 2024, which does not overlap with the pre-training data of LucaOne (before 29 May 2022). This dataset focused on the lowest-tier GO annotations within the hierarchical annotation framework of the biological-processes subset, identifying the 12 most prevalent terms at this foundational level. Each GO term randomly samples 100 sequences between 100 and 2,500 amino acids in length, resulting in 1,200 protein sequences across the 12 GO terms (Supplementary Note 2).
Convergence of nucleic acid and protein sequences for the same gene: we prepared an additional dataset comprising nucleic acid and protein sequences for the same genes and examined their correlations solely on embeddings. The results indicated that, despite nucleic acid and protein sequences not being paired during model training, those corresponding to the same gene demonstrated convergence within the LucaOne Embedding Space. More details in Supplementary Note 6 and Supplementary Fig. 12.
Task on pseudogene correction: we conducted a mask task prediction analysis (zero shot) on the data of the true gene (protein coding) and pseudogene pairs. The higher pseudogene correction rate and the true gene recovery rate demonstrated the model’s ability to identify the differences between pseudogenes and functional genes. More details in Supplementary Note 7, and Supplementary Figs. 13 and 14.
Task on codon degeneracy: we designed an additional task based on influenza virus haemagglutinin sequence data to verify whether LucaOne can distinguish between synonymous and non-synonymous mutations in a zero-shot manner (more details in Supplementary Fig. 16).
Details of central dogma tasks
Dataset construction, original dataset: we devised an experimental task to determine whether LucaOne has established the intrinsic association between DNA sequences and their corresponding proteins. A total of 8,533 accurate DNA–protein pairs from 13 species were selected in the NCBI RefSeq database, each DNA sequence extending to include an additional 100 nucleotides in the 5′ and 3′ contexts, preserving intron sequences within the data. In contrast, we generated double the number of negative samples by implementing substitutions, insertions and deletions within the DNA sequences or altering amino acids in the protein sequences to ensure the resultant DNA sequences could not be accurately translated into their respective proteins, resulting in a total of 25,600 samples—DNA–protein pairs. Then the positive and negative samples were each subjected to random shuffles and subsequently divided into 32 equally sized subsets. Then these subsets were assigned to the training, validation and testing sets in a 4:3:25 ratio. For more details, see Extended Data Table 4 and ‘Data availability’.
Analysis of misclassified samples: we analyse the misidentified samples from two perspectives—sequence and embedding. The relationship between sequence identity metrics and the prediction accuracy of the LucaOne embedding is presented in Extended Data Fig. 4a,b. Data details are presented in Supplementary Note 3. Extended Data Fig. 4a,b shows that the prediction accuracy of LucaOne for mutated sample pairs improved as sequence similarity decreased. We also evaluated the embedding distance alterations corresponding to modifications in nucleic acid and protein sequences by employing mean pooling to calculate these distances. As illustrated in Extended Data Fig. 4c,d, greater changes in embedding distances were correlated with improved predictive precision.
Dataset construction, two more species of urochordates: we incorporated two species with annotated reference genome urochordates (referred to as tunicate in the NCBI taxonomy) into our dataset: Styela clava (ASM1312258v2, GCF_013122585.1) and Oikopleura dioica (OKI2018_ I68_ 1.0, GCA_ 907165135.1). For each of these urochordate species, 480 genes were randomly selected, and positive gene samples, nucleic acid negative samples and protein-negative samples were constructed using the same approach as in the original dataset. The same data shuffling and partitioning principles were applied and integrated with the original dataset to retrain the central dogma model. Data details and model performance are presented in Extended Data Table 3, Extended Data Fig. 5 and ‘Data availability’.
Comparative performance analysis: upon integrating two additional urochordate species data, dataset version 2 as the model showed performance comparable to the original dataset models across all species except C. intestinalis. In particular, the F1 score for C. intestinalis improved significantly, despite the nearly unchanged accuracy. These findings suggest that supplementing the dataset with species that utilize a codon code similar to C. intestinalis enhances the model’s sensitivity to DNA–protein correlations in these organisms while preserving its sensitivity to DNA–protein correlations in species adhering to the standard codon code. For more details, see Extended Data Table 3 and ‘Data availability’.
CDS–protein task: in the current NCBI RefSeq database, genomes with complete intron annotations are limited, and the accuracy of intron predictions from alternative tools may directly impact model performance. To mitigate these challenges, coding sequence (CDS) regions corresponding to genes in the original dataset were extracted as intron-free nucleic acid sequences to perform the same task. See Supplementary Notes 4 for data details and Fig. 3d for analysis.
Task for cross-species homologous gene pairs: we designed an additional task related to the central dogma by modifying the negative samples in the original study. Instead of manually altering the sequences, the negative samples were replaced with homologous genes from closely related species. Please refer to Supplementary Notes 5 for details.
Downstream tasks details
Genus taxonomy annotation (GenusTax): this task is to predict which genus (taxonomy) the nucleic acid fragment may come from, which is very important in metagenomic analysis. A comparative dataset was constructed utilizing NCBI RefSeq, comprising 10,000 nucleic acid sequences, each extending 1,500 nucleotides and annotated with labels corresponding to 157 distinct genera (distributed as 33, 50, 29 and 45 across the four kingdoms: Archaea, Bacteria, Eukaryota and Viruses, respectively). The dataset was randomly segregated into training, validation and testing sets, adhering to an 8:1:1 partitioning ratio. It is important to note that while the LucaOne pre-training task utilized taxonomy annotations at the order level, the current task employs more granular genus-level annotations, thereby preventing label information contamination. This dataset was also employed for two additional analyses: predicting the taxonomy of sequences at the superkingdom and species levels. The details are presented in Extended Data Table 5.
Prokaryotic protein subcellular location (ProtLoc): this task is to predict the subcellular localization of proteins within prokaryotic cells, which has garnered substantial attention in proteomics due to its critical role50. It involves classifying proteins into one of six subcellular compartments: the cytoplasm, cytoplasmic membrane, periplasm, outer membrane, cell wall and surface, and extracellular space. Our approach adopted the same dataset partitioning strategy as DeepLocPro26, a model based on experimentally verified data from the UniProt and PSORTdb databases. For this dataset, we additionally designed a task based on the corresponding nucleic acid embeddings of the proteins. The result showed that embeddings derived from nucleic acid sequences are applicable to the task related to their corresponding protein sequences. The dataset and analytical results are provided in Supplementary Notes 8.
Protein stability (ProtStab): the evaluation of protein stability is paramount for elucidating the structural and functional characteristics of proteins, which aids in revealing the mechanisms through which proteins maintain their functionality in vivo and the circumstances that predispose them to denaturation or deleterious aggregation. We utilized the same dataset from TAPE51, which includes a range of de novo-designed proteins, natural proteins, mutants and their respective stability measurements. It is a regression task; each protein input (x) correlates with a numerical label (y ∈ ℜ), quantifying the protein’s intrinsic stability.
Non-coding RNA family (ncRNAFam): ncRNA represents gene sequences that do not code for proteins but have essential functional and biological roles. The objective is to assign ncRNA sequences to their respective families based on their characteristics. For this purpose, we utilize the dataset from the nRC52, which is consistent with the data employed in the RNAGCN53 study. Our methodology adheres to the same data partitioning into training, validation and testing sets as done in these previous studies, enabling direct comparison of results. This project involves a multi-class classification challenge that encompasses 88 distinct categories.
Influenza A antigenic relationship prediction (InfA): one of the foremost tasks in influenza vaccine strain selection is monitoring haemagglutinin variant emergence, which induces changes in the virus’s antigenicity. Precisely predicting antigenic responses to novel influenza strains is crucial for developing effective vaccines and preventing outbreaks. The study utilizes data from the PREDAC54 project to inform vaccine strain recommendations. Each data pair in this study comprises two RNA sequences of the haemagglutinin fragment from distinct influenza strains, accompanied by corresponding antigenic relationship data. The objective is framed as a binary classification task identifying the antigenic similarity or difference between virus pairs.
Protein–protein interaction (PPI): the forecasting of PPI networks represents a significant area of research interest. Our study utilized the DeepPPI55 database, whose positive dataset samples were sourced from the Human Protein Reference Database after excluding redundant interactions, leaving 36,630 unique pairs. This dataset was randomly partitioned into three subsets: training (80%), validation (10%) and testing (10%). The primary objective of this research is to perform binary classification of PPI sequences.
ncRNA–protein interactions (ncRPIs): an increasing number of functional ncRNAs, such as snRNAs, snoRNAs, miRNAs and lncRNAs, have been discovered. ncRNAs have a crucial role in many biological processes. Experimentally identifying ncRPIs is typically expensive and time-consuming. Consequently, numerous computational methods have been developed as alternative approaches. For comparison, we have utilized the same dataset as the currently best-performing study, ncRPI-LGAT27. It is a binary classification task involving pairs of sequences.
Comparison result details
We conducted a series of comparative experiments. According to Fig. 4, for all embedding methods, we compare whether the transformer encoder and two pooling strategies (max pooling and value-level attention pooling) were used on the model. At the hyperparameter level, we compared the number of encoder layers with the number of heads (4 layers with 8 heads and 2 layers with 4 heads), the peak learning rate of the Warmup strategy (1 × 10−4 and 2 × 10−4), and the batch size (8 and 16). Extended Data Table 5 shows the result of comparing whether the encoder was used and which pooling method was used accordingly, and Supplementary Fig. 4 shows more metrics on comparison experiments.
In the ProtLoc task, LucaOne’s accuracy is very close to that of the ESM2-3B. In the ncRPI task, the accuracy of the simple network with LucaOne’s embedding matrix is less than that of ncRPI-LGAT27 but higher than that of DNABert2 + ESM2-3B. In the other five tasks, we achieved the best results. It is better not to use an encoder for ProtLoc, InfA, PPI and ncRPI tasks. Using the max pooling strategy straightforwardly for the ncRNAFam and GenusTax tasks can obtain better results. We extended 2 tasks, 4 superkingdoms and 180 species prediction tasks for the genus classification task with the same sequence data. LucaOne’s accuracy improved by 0.1 and 0.054, respectively. In particular, LucaOne is more effective than other large models in embedding sequences without an encoder.
Computational resources
The data processing and training operations for LucaOne were carried out on Alibaba Cloud Computing. In addition, several tasks related to further processing or downstream computing were performed on alternative computing platforms, including Yunqi Academy of Engineering (Hangzhou, China) and Zhejiang Lab (Hangzhou, China).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Don’t miss more hot News like this! AI/" target="_blank" rel="noopener">Click here to discover the latest in AI news!
2025-06-18 00:00:00